Neuropatía, por un diagnóstico más preciso
La neuropatía es un conjunto de trastornos que se producen cuando se daña el sistema nervioso periférico. La condición generalmente se conoce como neuropatía periférica, y el daño en los axones nerviosos genera dolor y entumecimiento en manos y pies. Puede ser consecuencia de lesiones traumáticas, infecciones, trastornos metabólicos y exposición a toxinas. Una de las causas más comunes es la diabetes.
La enfermedad puede afectar a los nervios que controlan el movimiento muscular (nervios motores) y los que detectan sensaciones tales como el frío o dolor (nervios sensoriales). En algunos casos - como la neuropatía autonómica - puede incluir a órganos internos, como el corazón, vasos sanguíneos, vejiga e intestinos.
Existe una gran variedad de tratamientos disponibles para la neuropatía periférica, que van desde medicamentos y cremas tradicionales hasta dietas y terapias especiales, las cuales estimulan el sistema nervioso. Los antidepresivos, especialmente los tricíclicos y los inhibidores selectivos de la recaptación de serotonina y noradrenalina, son ampliamente aceptados, ya que alivian el dolor neuropático en personas no deprimidas. Otra clase de medicamentos comúnmente recetados son los anticonvulsivos. Éstos bloquean los canales de calcio en las neuronas pudiendo reducir el dolor. Por último, también se utilizan opioides para el tratamiento, pero con bastante precaución debido al riesgo de dependencia, sin embargo, son más coherentes y eficaces en la reducción del dolor.
Microtúbulos, transporte axonal y neuropatía
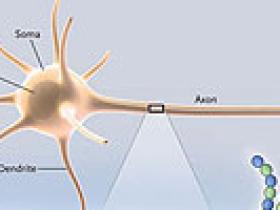
La dimensión física de una célula rara vez es su talón de Aquiles. Sin embargo, para las neuronas que se ven afectadas en la mayoría de los tipos de la neuropatía periférica, la longitud de sus axones es lo que mejor da cuenta de su vulnerabilidad selectiva. Como se muestra en la figura 1, el axón se ubica de forma contigua a su soma celular. El soma sintetiza los componentes de los axones y sus terminales, que pueden alcanzar hasta un metro de distancia. El transporte axonal es impulsado por motores moleculares a través de una "autopista" formada por microtúbulos. Las kinesinas conducen el transporte hacia el exterior, entregando nuevos bloques de construcción para los axones (por ejemplo, neurofilamentos) y terminales, llevando y trayendo orgánulos como las mitocondrias a zonas de alta demanda energética. El transporte desde los terminales del soma celular es impulsado por la dineína citoplasmática y su activador dinactina, y llevan orgánulos de degradación (lisosomas y autofagosomas) y endosomas que contienen las plataformas de señalización de los terminales.
Los microtúbulos se forman a partir de la asociación de los dímeros de α-tubulina y β-tubulina en protofilamentos, que se asocian lateralmente para formar un túbulo. La adición de subunidades de tubulina en el final de los filamentos conduce a un crecimiento del polímero, mientras que la pérdida de subunidades produce un acortamiento. En las células en división, esta remodelación es un proceso dinámico, pero en las neuronas, los microtúbulos de proteínas asociadas (por ejemplo, tau) frenan la dinámica de los microtúbulos. Si bien se reconoce la importancia de los microtúbulos y los motores que se mueven a lo largo de ellos, los recientes descubrimientos de mutaciones en los genes que codifican los microtúbulos y sus proteínas motoras no han dilucidado las principales interrogantes. Estas alteraciones causan enfermedades del neurodesarrollo y neurodegenerativas de diversos fenotipos, como la polimicrogiria asimétrica, el síndrome de Perry, un tipo de neuropatía motora, una forma de paraplejia espástica hereditaria, y varias formas de Charcot-Marie-Tooth, término para la neuropatía hereditaria.
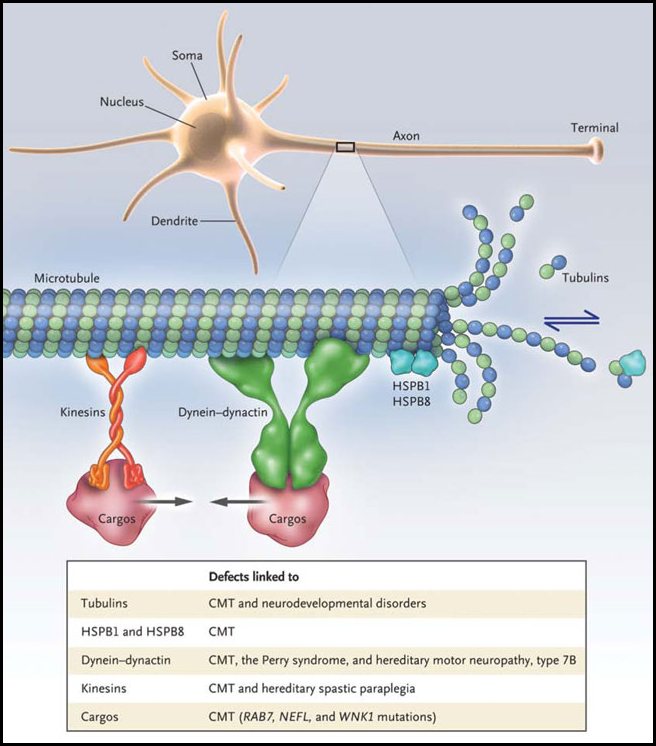
Los microtúbulos y sus proteínas motoras mantienen los axones. Los microtúbulos están formados por la polimerización de dímeros de α-tubulina y β-tubulina, que dinámicamente se ensamblan en una estructura polarizada que sirve como una pista para las proteínas motoras (dineína dinactina y kinesinas). Los defectos genéticos que afectan a la tubulina, dineína, dinactina, kinesinas, y los relacionados con Rab7 y la luz de neurofilamentos (NEFL) están ligados con la enfermedad Charcot-Marie-Tooth (CMT) y otras neuropatías. Un informe reciente de L. Almeida-Souza y colaboradores sugiere que HSPB1, una pequeña proteína de choque térmico que es mutante para la enfermedad CMT, también afecta la dinámica de los microtúbulos y por lo tanto, la salud de los axones.
Las mutaciones dominantes de HSPB1 y HSPB8 también causan una neuropatía axonal hereditaria. Estos genes codifican miembros de la familia de las pequeñas proteínas de choque térmico (sHSPs), todas los cuales se unen a proteínas desplegadas y evitan su agregación. A pesar de la expresión generalizada de HSPB1 y HSPB8, la neuropatía axonal predominante motora es el único fenotipo de estas mutaciones dominantes. Sobre la base de un trabajo anterior, que había demostrado que un subgrupo de mutantes dominantes HSPB1 mejoran la unión a las proteínas de sus clientes, Leonardo Almeida-Souza y colaboradores (J Neurosci 2011; 31:15320-15328) han concluido recientemente que la tubulina es una pareja de unión de HSPB1. Con el uso de ensayos bioquímicos y celulares, los autores demostraron que algunos mutantes HSPB1 potencian la unión a la tubulina, mejoran la estabilidad y dinámica de los microtúbulos. Además, propusieron que la estabilización de los microtúbulos es el mecanismo por el cual las proteínas HSPB1 mutantes dominantes causan una neuropatía dependiente de la longitud.
La idea tiene un paralelo interesante con respecto al paclitaxel, un fármaco quimioterapéutico que puede causar neuropatía. El paclitaxel se une a los microtúbulos con alta afinidad, lo que lleva a la estabilización, un efecto similar al causado por unas HSPB1 dominantes mutantes. Ya que los microtúbulos estabilizados quedan marcados para los motores de microtúbulos, no está claro cómo la modesta estabilización observada conduce a la degeneración axonal. Una posibilidad es que esta estabilización bloquee la remodelación del citoesqueleto en los sitios sinápticos. Por otra parte, la unión de mutantes HSPB1 a lo largo de los microtúbulos podría impedir el transporte; en particular la movilidad de la kinesina que es susceptible a este efecto.
Es poco probable sin embargo, que los microtúbulos estabilizados sean la respuesta definitiva a la cuestión de cómo los mutantes HSPB1 causan neuropatía. No todos los HSPB1 mutantes afectan a la actividad de las chaperonas. El sHSPs se une a muchos sustratos, y hay otros efectos reportados de mutantes SHSP. Por ejemplo, la sobreexpresión de los dos diferentes HSPB1 mutantes (pero no de HSPB1 de tipo salvaje) genera la agregación de HSPB1 junto con otras proteínas, incluyendo las subunidades de neurofilamentos. Estos agregados podrían ser tomados como evidencia que el HSPB1 mutante tiene minimizada su actividad de acompañante, por lo menos hacia estos sustratos. Otra cuestión que hace que la elucidación del mecanismo sea particularmente difícil es que si una neuropatía tarda años en desarrollarse, se esperaría que los mutantes que la causan tengan defectos relativamente sutiles (demostrado recientemente con una versión mutante específica para Rab7, que causa la enfermedad de Charcot-Marie-Tooth). Sólo con el tiempo se podrán demostrar los efectos acumulativos de la mutación sobre la función celular.
Fuente bibliográfica
Microtubules, Axonal Transport, and Neuropathy
Erika L.F. Holzbaur, Ph.D., and Steven S. Scherer, M.D., Ph.D.
Departments of Physiology (E.L.F.H.) and Neurology (S.S.S.), Perelman School of Medicine at the University of Pennsylvania, Philadelphia.
N Engl J Med 2011; 365:2330-2332