Corrección genética en bebé con enfermedad congénita
El uso personalizado de la técnica de CRISPR en un lactante con hiperamonemia, una rara condición metabólica, muestra efectos prometedores y abre nuevas posibilidades para tratamientos duraderos de patologías graves y poco frecuentes.
Un estudio innovador publicado en The New England Journal of Medicine, describe un enfoque personalizado de edición genética para tratar un error congénito del metabolismo, específicamente la deficiencia de carbamoil-fosfato sintetasa 1 (CPS1). La CPS1 es una enzima mitocondrial esencial para el ciclo de la urea, que cataliza la conversión de amoníaco y bicarbonato en carbamoil fosfato. La deficiencia de esta enzima provoca una acumulación tóxica de amoníaco en la sangre, resultando en hiperamonemia, una condición que puede causar daño cerebral severo, coma y muerte si no se trata.
Las intervenciones clínicas actuales, como la diálisis, los secuestradores de amoníaco, la restricción de proteínas y el trasplante de hígado, tienen limitaciones significativas y a menudo no logran prevenir resultados neurológicos adversos.
El objetivo principal de la investigación, liderada por el Dr. Kiran Musunuru de la Universidad de Pensilvania, EE. UU., fue evaluar la viabilidad y eficacia de una terapia de edición genética personalizada para corregir la deficiencia de CPS1 en un lactante afectado. El estudio fue realizado en el Center for Experimental Neurotherapeutics, St. Jude Children's Research Hospital. Se buscaba demostrar la corrección del defecto genético subyacente en lugar de simplemente mitigar los síntomas, ofreciendo una posible cura para esta enfermedad rara y grave.
Los investigadores utilizaron una técnica de edición de bases de adenina (ABE). Esta estrategia permite la conversión precisa de un par de bases adenina-timina (A-T) a un par de bases guanina-citosina (G-C) en el genoma. El enfoque implicó la creación de un editor de bases ABE personalizado, con un ARN guía (gRNA), diseñado para dirigirse a la mutación específica del paciente en el gen CPS1.
La enzima Cas9 nickase (Cas9n) fue utilizada para crear una hendidura en la cadena de ADN, lo que permite a la desaminasa de adenosina convertir la adenina mutante en inosina, que luego se empareja con la citosina, corrigiendo así la mutación (figura 1). Para acelerar el proceso de personalización del editor genético, se insertó un segmento del gen CPS1 mutado del paciente en una línea celular de hepatocitos humanos para facilitar las pruebas de combinaciones ABE:gRNA.
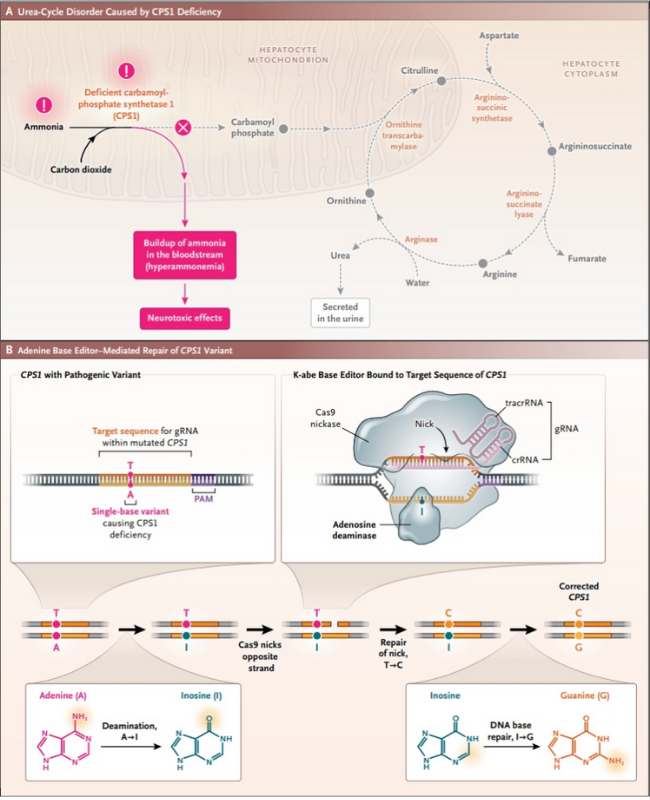
Figura 1: estrategia para tratar la deficiencia de CPS1 mediante la edición de bases
Los resultados preclínicos demostraron que el editor de bases ABE fue capaz de corregir eficientemente la mutación CPS1 en las células del paciente in vitro. Además, los estudios en modelos de ratón transgénicos y primates no humanos mostraron la eficacia y la relativa seguridad de la terapia. Tras estos resultados prometedores, se administró el editor genético personalizado al lactante afectado.
Aunque los autores notaron una estabilización clínica a corto plazo, la ausencia de confirmación molecular directa de la edición genética a través de una biopsia hepática deja preguntas sobre la durabilidad del efecto terapéutico, la extensión del mosaicismo de la edición y los riesgos de eventos fuera del objetivo o respuestas inmunitarias.
El estudio concluye que la edición genética personalizada es una estrategia terapéutica prometedora para enfermedades genéticas raras como la deficiencia de CPS1. La capacidad de intervenir a nivel genómico y corregir directamente el defecto genético subyacente en el hígado del paciente representa un avance significativo en la medicina basada en CRISPR. Sin embargo, los autores señalan que se trata de una experiencia n=1 en una condición ultra rara, lo que limita la generalización de los resultados.
Además, enfatizan la necesidad de un seguimiento a largo plazo del paciente tratado para evaluar la durabilidad del efecto terapéutico y la seguridad de la terapia. También sugieren el desarrollo de biomarcadores mínimamente invasivos en lugar de biopsias hepáticas para evaluar el éxito de la edición y la expansión a cohortes pequeñas y bien controladas. Se propone la consideración de estudios de ureagénesis utilizando trazadores de isótopos estables, ensayos de ADN libre de células o metabolómica cuantitativa.
Asimismo, se destaca la importancia de la personalización continua de las plataformas de administración y la mejora de la eficiencia de la edición para traducir este éxito individual en una terapia reproducible y escalable. Por último, se subraya la necesidad de una gestión ética y regulatoria cuidadosa a medida que este campo avanza hacia la atención estándar.
Fuente bibliográfica
Personalized Gene Editing to Treat an Inborn Error of Metabolism
Andrea L. Gropman, M.D., and Alexis C. Komor, Ph.D.
Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis
DOI: 10.1056/NEJMe2505721