Tratando una enfermedad hepática grave
La forma clásica de la deficiencia de alfa1-antitripsina (AT) es provocada por una mutación puntual (sustitución de lisina por ácido glutámico en el residuo 342) que altera el plegamiento de una glicoproteína plasmática derivada del hígado durante la biogénesis y que también estimula la polimerización. Además de la formación de agregados insolubles en el retículo endoplásmico de las células hepáticas, existe una reducción del 85 al 90% en las concentraciones circulantes de AT, el principal inhibidor fisiológico de la elastasa de neutrófilos.
Por otra parte, la fibrosis hepática y la carcinogénesis son provocadas por un mecanismo de ganancia de tóxicos, de hecho, la deficiencia de AT es la causa genética más común de enfermedad hepática en la infancia pero también se puede presentar por primera vez con cirrosis y/o carcinoma hepatocelular en la edad adulta.
Deficiencia de alfa1-antitripsina y autofagia
La deficiencia del alelo de SERPINA1 (PI*Z), que codifica el inhibidor de la serina proteasa (PI) alfa1-antitripsina, da lugar a la formación de inclusiones intrahepáticas que causan la cirrosis. La única terapia curativa para los pacientes con enfermedad hepática grave es el trasplante. Tunda Hidvegi y colaboradores acaban de describir un nuevo método para eliminar las inclusiones patológicas en modelos celulares y animales de la enfermedad hepática como consecuencia de la mutación Z. Los autores utilizaron el fármaco antiepiléptico carbamazepina para estimular la "autodigestión celular", o autofagia (Science 2010; 329(5988):229-232). La pregunta es si este método será útil para el tratamiento de la enfermedad hepática asociada con la deficiencia de alfa1-antitripsina en seres humanos y si también sería eficaz en el manejo de otras condiciones, como la enfermedad de Huntington y el Parkinson, que también son resultado de la retención intracelular de las proteínas mutantes.
La alfa1-antitripsina se sintetiza predominantemente en el hígado y es secretada a la circulación, donde controla la degradación de los tejidos por la enzima elastasa de neutrófilos. La deficiencia del alelo Z se encuentra en el 4% de las personas blancas del norte de Europa, aproximadamente 1 de cada 2.500 son homocigotas para el alelo. Se estima que unos 100.000 individuos son portadores de PI*Z en los Estados Unidos. La mutación hace que la cadena polipeptídica de alfa1-antitripsina recién sintetizada se pliegue inadecuadamente dentro de los hepatocitos y por lo tanto sea objeto de la degradación por autofagia y por el proteasoma - esto último por medio de un proceso denominado degradación asociada al retículo endoplasmático (figura 1). Las proteínas que escapan de la degradación forman cadenas de polímeros que se unen entre el centro reactivo de una molécula y la hoja beta A de otra. Estos polímeros tipo "hojas de bucle", junto con las moléculas solubles, se conservan en el retículo endoplasmático de los hepatocitos, su acumulación se asocia con la activación del factor nuclear-kB, el cual puede conducir a la proliferación celular, la activación de las caspasas, y en última instancia, a la muerte celular. Estos procesos celulares pueden ser la base de patologías hepáticas tales como la hepatitis neonatal, la cirrosis y el carcinoma hepatocelular. La falta de concentraciones circulantes alfa1-antitripsina expone a los pulmones al ataque proteolítico por la elastasa de neutrófilos y por tanto, predispone a los homocigotos PI*Z al inicio precoz del enfisema.
Una comprensión detallada de la relación proteína y los polímeros patológicos de alfa 1-antitripsina ha llevado al desarrollo de una serie de estrategias terapéuticas. La polimerización de alfa1-antitripsina Z puede ser bloqueada in vitro por péptidos y pequeñas moléculas, pero esto no se ha evaluado en modelos animales para la enfermedad. Chaperones químicos que estabilizan la proteína monomérica también son eficaces in vitro y en animales, pero tienen que someterse a una prueba rigurosa de eficacia en personas con deficiencia de alfa1-antitripsina.
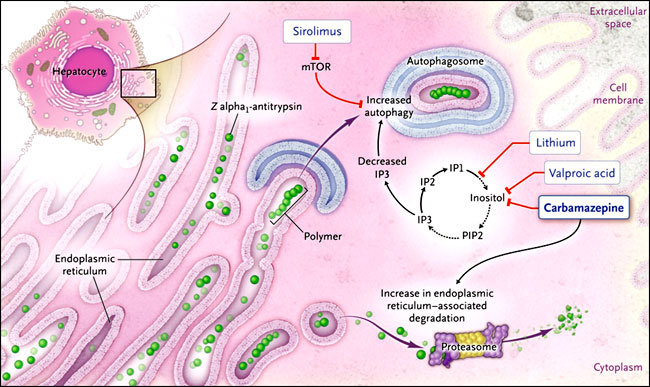
La alfa1-antitripsina Z mutante es degradada por los proteasomas o formas de polímeros que se conservan en el retículo endoplásmico de los hepatocitos. El grupo de Tunda Hidvegi adoptó un nuevo enfoque mediante el uso de un medicamento aprobado por la FDA, la carbamazepina (otras posibilidades se indican en las casillas blancas), para mejorar la degradación de los mutantes alfa1-antitripsina por el proteasoma (por medio de la degradación endoplásmica asociada al retículo) y estimular la autofagia para eliminar los polímeros en hígados de ratones. La carbamazepina reduce la concentración de inositol libre en el hepatocito y por lo tanto estimula la autofagia de los polímeros alfa1-antitripsina Z por medio de una vía independiente de la rapamicina de los mamíferos (mTOR). El inositol es necesario para constituir el fosfatidilinositol-4,5-bifosfato (PIP2), que a su vez se metaboliza a inositol trifosfato (IP3), inositol bifosfato (IP2) y el monofosfato de inositol (IP1). Se cree que la disminución de IP3 sería responsable de la activación de la autofagia.
Estudios previos han demostrado que la autofagia contribuye a la eliminación de los mutantes alfa1-antitripsina. T Hidvegi y colegas evaluaron el efecto de los agentes pro-autofágicos en modelos de la enfermedad. Eligieron el agente antiepiléptico carbamazepina de una lista de medicamentos que mejoran la degradación autofágica de las proteínas con largos tramos de repeticiones de poliglutamina, tales unidades también forman agregados celulares. Los autores eligieron la carbamazepina porque tiene un gran perfil de seguridad en seres humanos. Encontraron que el medicamento no sólo aumenta la autofagia, sino también la eliminación proteosomal de la alfa1-antitripsina Z soluble e insoluble en las líneas celulares (figura 1). El hallazgo más sorprendente fue en ratones que expresan el mutante de alfa1-antitripsina Z. Estos animales acumulan los polímeros en inclusiones para el ácido periódico-Schiff en el hígado, una característica de la enfermedad humana. En ratones machos jóvenes, el tratamiento con altas dosis de carbamazepina (10 a 20 veces las dosis utilizadas para tratar a pacientes con epilepsia) eliminó las inclusiones positivas de alfa1-antitripsina para el ácido periódico-Schiff dentro de 2 semanas. También hubo una reversión de la fibrosis hepática.
Estos resultados proporcionan un fuerte apoyo a la realización de ensayo clínicos randomizados y controlados para evaluar la eficacia de la carbamazepina en pacientes con enfermedad hepática como consecuencia de la deficiencia de alfa1-antitripsina. Sería importante determinar si los efectos observados en ratones pueden ser recapitulados en dosis mucho más bajas que puedan ser administradas en los seres humanos. La reducción de las inclusiones hepáticas y la disminución de la progresión de la enfermedad hepática es más sencilla que la evaluación de la eficacia de los agentes pro-autofágicos en personas con enfermedades neurodegenerativas, como la enfermedad de Huntington y la enfermedad de Parkinson, que se derivan de las inclusiones intracelulares de la huntingtina y la alfa-sinucleína, respectivamente. En caso de ensayos con resultados negativos, todavía habría una posibilidad para evaluar otros medicamentos aprobados por la Administración de Alimentos y Medicamentos (FDA) con efectos pro-autofágicos en modelos celulares y animales para la enfermedad, específicamente enfocados a cualquiera de las vías dependientes de la rapamicina en mamíferos (mTOR) (por ejemplo, sirolimus) o las vías independientes de mTOR (por ejemplo, el litio y el ácido valproico) (figura 1).
Fuente bibliográfica
Alpha1-Antitrypsin Deficiency and Autophagy
Stefan J. Marciniak, Ph.D., M.R.C.P., and David A. Lomas, Ph.D., F.R.C.P.
Department of Medicine, University of Cambridge, Cambridge Institute for Medical Research, Cambridge, United Kingdom.
N Engl J Med 2010; 363(19):1863-1864