Depleción de linfocitos, clave para la virulencia
Las secuencias genómicas del SARS-CoV-2, reunidas a partir de 112 muestras de alta calidad junto con las secuencias del conjunto de datos de la Iniciativa mundial para el intercambio de datos sobre la influenza (GISAID), muestran una evolución estable y sugieren que existen dos linajes principales con un historial de exposición diferencial durante la fase inicial del brote en Wuhan. No obstante, estos aislados exhibieron una virulencia y resultados clínicos similares. La linfocitopenia, especialmente la reducción de los recuentos de células T CD4+ y CD8+ en el momento de la admisión hospitalaria, se catalogó como predictiva de la progresión de la enfermedad. Se observaron altos niveles de interleucina (IL)-6 y de IL-8 durante el tratamiento en pacientes con enfermedad grave o crítica y se correlacionaron con la disminución del recuento de linfocitos. Los factores determinantes de la severidad de la patología parecían derivarse principalmente de factores del hospedero como la edad y la linfocitopenia (y la tormenta de citoquinas asociada), mientras que las variaciones genéticas virales no afectaban significativamente los resultados.
Linajes virales
El coronavirus 2 del síndrome respiratorio agudo severo (SARS-CoV-2) surgió a finales de 2019, y ciertos aspectos de la enfermedad que causa -COVID-19- siguen desconcertando a clínicos e investigadores. Se estima que el SARS-CoV-2 ya ha infectado a más de 9 millones de personas y ha cobrado más de 450.000 vidas en todo el mundo, y esta pandemia ha paralizado las economías a nivel mundial. En un reciente artículo publicado en Nature, Xiaonan Zhang y colaboradores del Centro Clínico de Salud Pública de Shanghai presentan datos sobre la evolución de dos grandes linajes de SARS-CoV-2, junto con información relativa a los determinantes de la gravedad de la enfermedad, a partir de su análisis de 326 personas infectadas en Shanghai (China) (DOI: 10.1038/s41586-020-2355-0).
Se pensó inicialmente que el SARS-CoV-2, que tomó al mundo por sorpresa, había "saltado" a los humanos desde un animal en el Mercado Mayorista de Mariscos de Huanan en Wuhan, China. Cuando los primeros casos de una enfermedad previamente desconocida, inicialmente descrita como "una neumonía severa de etiología desconocida", fueron identificados en Wuhan a finales de diciembre de 2019, la mayoría de los casos pudieron ser rastreados hasta este mercado. La implicación era que el nuevo coronavirus había cruzado la barrera de las especies en el mercado a partir de un animal en venta vivo infectado. El pangolín malayo, un oso hormiguero escamoso que antes vivía en relativa oscuridad, se enfrentó repentinamente a acusaciones de ser el culpable, aunque no se sabe con certeza si esta criatura protegida estaba a la venta en el mercado en ese momento. Sin embargo, algunos casos de la enfermedad a principios de diciembre de 2019 en Wuhan no tenían vínculos evidentes con el mercado.
Zhang y sus colegas analizaron 94 secuencias completas del genoma del SARS-CoV-2 en muestras obtenidas de personas que vivían en Shangai y que habían visitado un centro hospitalario en enero o febrero de 2020, y compararon esos datos con otras 221 secuencias del virus. Los resultados de los autores refuerzan observaciones previas de dos importantes linajes filogenéticos (clados) del SARS-CoV-2 durante la fase inicial del brote en China (DOI: 10.1093/nsr/nwaa036). Se distinguen por dos diferencias de nucleótidos distintivas, que sugieren múltiples orígenes para las infecciones humanas transmitidas a personas en Shanghai (que está a unos 800 kilómetros por carretera de Wuhan).
Los dos linajes se denominan clados I y II (figura 1). Presumiblemente evolucionaron independientemente de un ancestro común, pero su ascendencia en términos de cómo se relacionan entre sí no está clara, porque solo difieren en dos sitios genómicos. Una de las diferencias se refiere a un nucleótido particular en la secuencia que codifica el residuo de aminoácido número 84 en la proteína viral ORF8. Si el nucleótido tiene una base de timina (clado I), la secuencia codifica el aminoácido leucina; si tiene una base de citosina (clado II), la secuencia codifica una serina. La otra diferencia está en un nucleótido del gen ORF1ab, que contiene citosina (clado I) o timina (clado II); ambas secuencias de nucleótidos resultantes codifican la serina.
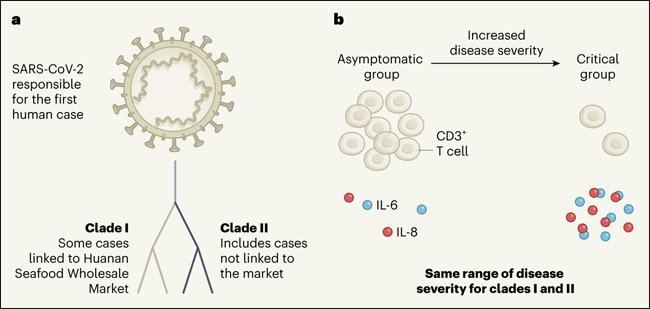
Figura 1 | Evaluando la relación entre los linajes del coronavirus y la gravedad de COVID-19.
Zhang y sus colegas estudiaron a personas de Shangai (China) que se infectaron con el coronavirus SARS-CoV-2 a principios de 2020. a, En concordancia con investigaciones anteriore, las secuencias del genoma del coronavirus que Zhang y colaboradores identificaron pertenecían a dos linajes, denominados clado I y clado II. Estos difieren en dos nucleótidos y probablemente evolucionaron independientemente de un ancestro común. El clado I se asoció con algunos casos relacionados con el mercado mayorista de mariscos de Huanan en Wuhan (China), que originalmente se pensaba que era la fuente del brote, mientras que los autores encontraron infecciones del clado II que no tenían vínculos con el mercado. Ambos linajes podrían haberse propagado independientemente al mismo tiempo. b, Zhang y sus colegas clasificaron a los individuos en cuatro grupos, según la gravedad de su enfermedad, que iban desde los no afectados por los síntomas (el grupo asintomático) hasta el grupo crítico (los que necesitaban ventilación artificial para respirar). Ambos clados tenían la misma capacidad para causar los diferentes grupos de enfermedades. El aumento de la gravedad de la enfermedad se acompañó de un agotamiento de linfocitos T CD3+ y un aumento de las proteínas citoquinas proinflamatorias IL-6 e IL-8. Los altos niveles de citoquinas pueden causar una intensa respuesta inmunológica conocida como tormenta de citoquinas.
Combinando la genómica viral con pruebas epidemiológicas de cómo las personas podrían haber contraído la infección, Zhang y sus colegas muestran que los genomas virales de seis personas con vínculos establecidos con el grupo de mercado de Wuhan en el clado I en el árbol familiar del SARS-CoV-2, mientras que los genomas virales de tres casos sin vínculos conocidos con el grupo de mercado en el clado II. Estos datos apoyan la idea de que el mercado podría no haber sido el origen de la pandemia. En cambio, sugieren que los clados I y II se originaron de un antepasado viral común y se propagaron independientemente al mismo tiempo: el clado I a través del mercado y el clado II fuera de él. Por lo tanto, la transferencia de animal a humano podría haber ocurrido en otro lugar, sembrando cadenas de transmisión que encontraron su camino hacia el mercado - donde la alta densidad de puestos y humanos susceptibles facilitó una propagación incontenible en el sitio y posteriormente más allá de él.
La circulación de los diferentes "tipos" de SARS-CoV-2 ha sido un tema polémico, derivado de la observación de distintos linajes filogenéticos. Sin embargo, se espera que esa divergencia genética entre los virus, especialmente en el contexto de los hospederos humanos "inmunológicamente ingenuos" (los que nunca antes se han encontrado con el virus). Esto puede explicarse por el "efecto fundador", que es común durante los brotes virales: si un número limitado de variantes virales entran aleatoriamente en una nueva región geográfica donde hay una población susceptible, su posterior propagación allí facilita el predominio de esas variantes en ese lugar.
Sin embargo, la diferencia de prevalencia de esas variantes en esa población particular, en comparación con las poblaciones infectadas de otras regiones, no equivale necesariamente a una mejora de la aptitud de esas variantes en lo que respecta a la replicación y la transmisión del virus. En consonancia con esta idea, Zhang y sus colegas no encuentran pruebas de ninguna asociación entre ninguno de los dos clados, ni entre ninguna mutación en los subclados, y los parámetros clínicos que evaluaron para categorizar la gravedad de la enfermedad COVID-19. Aunque este hallazgo no es sorprendente, dado que los dos clados difieren por solo dos nucleótidos de los aproximadamente 30.000 nucleótidos del genoma del SARS-CoV-2, pone de relieve el hecho de que los distintos linajes filogenéticos no indican necesariamente cepas virales distintas con resultados de enfermedad diferentes.
Al no haber encontrado ninguna diferencia en los resultados clínicos entre las infecciones con los dos linajes del SARS-CoV-2, los investigadores analizaron diversos parámetros de la función del sistema inmunológico en los hospederos humanos para identificar los factores que contribuyen a la gravedad de la enfermedad.
Los autores se centraron en cuatro categorías de enfermedades con definiciones bien descritas de los resultados clínicos. Los individuos menos afectados eran asintomáticos y no presentaban fiebre, ni problemas respiratorios ni signos de daño pulmonar en las radiografías. Los casos leves eran los de personas que tenían fiebre y signos de inflamación en las radiografías de sus pulmones, lo que indicaba una neumonía. Las personas con enfermedades graves tenían dificultad para respirar y presentaban signos de daño pulmonar descritos como "opacidades de vidrio esmerilado" en las radiografías. Los pacientes en estado crítico tenían el síndrome de dificultad respiratoria aguda y necesitaban ventilación mecánica. De acuerdo con investigaciones anteriores, Zhang y sus colegas encontraron que el hecho de ser mayor, la presencia de otras condiciones médicas preexistentes (comorbilidades), y el género masculino eran los principales factores asociados con una mayor probabilidad de enfermedades más graves.
A partir del análisis de las muestras de sangre, los autores proporcionan pruebas de los cambios que caracterizaron los casos graves y críticos de COVID-19. Una característica de estos casos fue la linfocitopenia, un número anormalmente bajo de linfocitos en sangre. Los investigadores atribuyeron esta linfocitopenia a la depleción de un tipo celular específico, el linfocito T CD3+, que probablemente refleja el movimiento de estas células desde la sangre a los sitios de infección en los tejidos.
Otra característica de los casos severos y críticos fueron los niveles anormalmente altos de las citoquinas IL-6 e IL-8, pequeñas proteínas que promueven la inflamación. Los altos niveles de citoquinas proinflamatorias impulsan una intensa respuesta inmunológica que se conoce comúnmente como tormenta de citoquinas. Las células del sistema inmunológico llamadas macrófagos, que están presentes en el pulmón, pueden producir IL-6 e IL-8, y a menudo son los mediadores celulares iniciales de una tormenta de citoquinas en otras infecciones respiratorias.
Sin embargo, aún no se han definido las poblaciones celulares precisas que contribuyen a la prolongada tormenta de citoquinas que se produce en algunos casos de COVID-19.
La correlación inversa entre los altos niveles de IL-6 o IL-8 y los bajos números de linfocitos apunta a los mecanismos subyacentes que podrían vincular estas características de la enfermedad grave. La posibilidad de que los altos niveles de citoquinas causen linfocitopenia es coherente con la observación de que las personas con COVID-19 que fueron tratadas con el fármaco tocilizumab, que bloquea la señalización mediada por la IL-6, recuperaron sus niveles de linfocitos en el torrente sanguíneo. Sin embargo, se necesitan más estudios experimentales y sobre los mecanismos involucrados para establecer si una conexión causal subyace a la correlación entre estos niveles de citoquinas y la linfocitopenia. Cabe destacar el marco temporal discordante de los cambios en estos dos parámetros: el agotamiento de las células T es evidente desde la primera semana de la enfermedad manifiesta, mientras que una tormenta de citoquinas surge más tarde, cuando COVID-19 se ha vuelto grave.
Además, ni la linfocitopenia ni la tormenta de citoquinas son exclusivas de COVID-19. Ambos son distintivos de muchos tipos de infección respiratoria grave, incluida la infección humana por los virus de la gripe aviar, y el síndrome respiratorio agudo severo (SARS), una enfermedad causada por un coronavirus relacionado con el SARS-CoV-2. Para delinear las firmas inmunológicas específicas de COVID-19, se necesitarán análisis celulares y moleculares más detallados.
El seguimiento de la evolución del SARS-CoV-2 es fundamental para informar las políticas de salud pública necesarias y limitar la propagación de las enfermedades. La disección de las causas y los mecanismos subyacentes de las defensas inmunitarias perturbadas, como la depleción de los linfocitos T CD3+ y el aumento de la respuesta proinflamatoria, así como la determinación de las firmas clínicas y moleculares cruciales de la COVID-19, son de importancia capital para el diseño de estrategias terapéuticas y vacunas eficaces. Zhang y colaboradores establecen algunas bases esenciales que deberían ayudar en estas grandes tareas, y su trabajo plantea preguntas clave que deberán ser respondidas si queremos limitar esta pandemia e intentar prevenir una futura.
Fuente bibliográfica
A race to determine what drives COVID-19 severity
Marios Koutsakos & Katherine Kedzierska
Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Parkville, Victoria 3010, Australia.
DOI: 10.1038/d41586-020-01915-3