Gen sustituto para la distrofia muscular
El producto esencial del gen de la distrofia muscular de Duchenne (DMD) es la distrofina, una proteína en forma de varilla que protege a los miocitos estriados de las lesiones inducidas por la contracción. La utrofina, una proteína relacionada, recapitula la mayoría de los elementos estructurales y de unión de la distrofina. En un nuevo estudio se diseñó un constructo génico optimizado que codifica una utrofina miniaturizada (µUtro), que se libera mediante vectores virales adeno asociados (AAV). Se demostró que µUtro es un sustituto altamente funcional y no inmunogénico de la distrofina, que previene los aspectos histológicos y fisiológicos más deletéreos de la distrofia muscular en modelos de animales pequeños y grandes. Tras la administración sistémica de un AAV-µUtro a ratones modelo de la enfermedad con déficit de distrofina neonatal, los marcadores histológicos y bioquímicos de mionecrosis y regeneración se suprimen completamente durante todo el crecimiento hasta el peso adulto. Además, en el modelo de golden retriever con deficiencia de distrofina, µUtro evitó de forma no tóxica la mionecrosis, incluso en los músculos más potentes. Estos hallazgos respaldan un modelo en el que las terapias derivadas de la utrofina podrían utilizarse para tratar la deficiencia clínica de distrofina, con un perfil inmunológico favorable.
Utrofina protectora
La distrofia muscular de Duchenne (DMD) es una enfermedad de desgaste muscular ligada al cromosoma X. Los niños afectados pierden la capacidad deambulatoria alrededor de los 12 años y fallecen de problemas respiratorios o cardíacos a los 20 o 30 años. La DMD es causada por mutaciones en el gen que codifica la distrofina, un enlace proteínico esencial entre el citoesqueleto interno y la matriz extracelular de las células musculares. Actualmente no hay un tratamiento efectivo. El progreso en los abordajes genéticos ha llevado a la aprobación de fármacos que promueven el salto del exón o que detienen la lectura del codón, pero la eficacia clínica necesita ser mejorada, y siendo específicos de la mutación, los fármacos aprobados no son efectivos en la mayoría de los individuos afectados. La terapia genética que utiliza el vector de virus adeno-asociado (AAV) es muy prometedora ya que trataría a todos los pacientes, independientemente de su mutación patogénica, introduciendo una versión funcional del gen de la distrofina en el músculo. En el estudio realizado por Song y colaboradores (DOI: 10.1038/s41591-019-0594-0) describen un AAV que contiene un gen truncado optimizado por el codón (µUtro) derivado de la proteína relacionada con la distrofina, la utrofina, y muestran que este constructo previene los síntomas distróficos en los modelos mdx de ratón y canino de la enfermedad en ausencia de una respuesta inmunológica.
Actualmente existen desafíos sustanciales para la terapia genética de la DMD usando AAV. El gen de la distrofina es demasiado grande para estos vectores virales, por lo que es necesario utilizar genes de microdistrofina truncados (µDys). Se necesitan altas dosis de este vector para expresar suficiente proteína para tener un efecto, y los pacientes deben ser preseleccionados para detectar anticuerpos que bloqueen la entrega del vector a los músculos. En un ensayo inicial, algunos pacientes mostraron una leve respuesta inmunológica a la distrofina cuando esta se expresó a partir del promotor del CMV activo (N. Engl. J. Med. 363, 1429–1437 (2010)). La administración de AAV-µDys en los ensayos de fase I en un pequeño número de pacientes mostró mejoras funcionales, pero más recientemente se han notificado algunos efectos adversos transitorios. Se han descrito varios diseños de µDys, pero todos se han modificado a partir de proteínas truncadas observadas en los pacientes más leves con distrofia muscular de Becker; por lo tanto, el tratamiento convertiría, en el mejor de los casos, la DMD en una BMD leve. El diseño de un gen truncado terapéutico que encajaría en el AAV, y maximizaría la función hacia lo normal mientras que minimizaría la respuesta inmune, podría potencialmente transformar este enfoque terapéutico.
La utrofina es un sustituto conocido de la distrofina, ya que su expresión en el modelo de ratón mdx para DMD previene la patología aunque carece de algunos dominios de interacción proteína-proteína presentes en la distrofina. Ni la eficacia funcional de la terapia génica con utrofina ni su inmunogenicidad se han probado nunca en animales grandes, lo que constituye una prueba más rigurosa para su uso en pacientes. Song y sus colegas hicieron predicciones sobre la forma óptima de acortar el dominio de la utrofina, que se ha demostrado que puede acomodar funcionalmente una variedad de eliminaciones. Lo hicieron utilizando estructuras cristalinas parciales de distrofina y utrofina, y teniendo en cuenta las supresiones del dominio de varillas en los parálogos de la utrofina a lo largo de la evolución. También optimizaron la secuencia acortada del ADN de la utrofina con codones conocidos por maximizar la expresión. El µUtro se parece más al gen de la microdistrofina deltaR4-R23/deltaCT actualmente en etapa de ensayo clínico (figura 1).
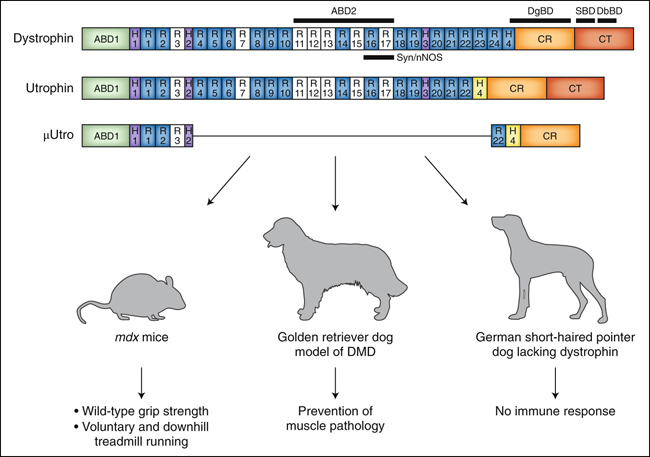
Figura 1. Terapia genética de la utrofina para la DMD.
Estructura de las proteínas de distrofina y utrofina clínicamente relevantes. La distrofina de longitud completa comprende el dominio N-terminal de unión a la actina (ABD), cuatro dominios de bisagra (H), 24 repeticiones similares a la espectrina (R) que forman el dominio "varilla", un ABD interno, el dominio de unión a la sintrofina alfa, que localiza la nNOS (Syn/nNOS), el dominio de unión de distroglicanos (DgBD), y los dominios de unión de sintrofinas y distrobrevinas (SBD, DbBD) localizados en los dominios ricos en cisteína (CR) y C-terminal (CT). Por debajo de la distrofina está la estructura de la utrofina, que tiene sólo 22 repeticiones similares a la espectrina y carece del dominio de unión a la sintrofina alfa. El vector AAV-µUtro se probó en el modelo de ratón mdx para la DMD, y en los modelos de perro golden retriever y de puntero de pelo corto alemán para la DMD.
Los autores administraron el AAV-µUtro bajo el control del promotor del CMV a ratones mdx neonatos antes de la aparición de la enfermedad como una prueba rigurosa de su eficacia. Su estudio reveló que el vector confería mioprotección durante todo el desarrollo muscular, hasta el final del estudio de cuatro meses, después de una única inyección intraperitoneal. Además, los ratones tratados con AAV-µUtro tenían una fuerza de agarre equivalente a la de los ratones silvestres y mostraban un comportamiento normal en la carrera voluntaria y en las bajadas. Esto no se había observado anteriormente utilizando µUtro o transgenes de utrofina de longitud completa: esos tratamientos previnieron la patología, pero no devolvieron la función completa a los niveles del tipo silvestre, aunque se ha observado con los vectores µDys de última generación en ratones mdx.
Aunque estos datos proporcionaron la esperanza de que el tratamiento con este µUtro diseñado podría proporcionar un fenotipo casi normal y no un fenotipo de DMD en los individuos afectados con DMD, la eficacia debe ser probada primero en un modelo canino de distrofia muscular del Golden Retriever (GRMD), ya que su fenotipo grave se parece más a los pacientes que los modelos de ratón. No observaron ninguna inmunidad celular contra el vector o el µUtro en perros neonatos, y se evitó la patología muscular. Dado que a los niños no se les suele diagnosticar la DMD antes de los dos años, Song y sus colegas también administraron el vector AAV-µUtro a perros jóvenes en altas dosis de vector junto con prednisona para reducir la inflamación. Los ejemplares tratados mostraron una completa supresión de la lesión muscular en curso. Sin embargo, dada la edad de los animales en el momento de la entrega del vector, cabe señalar que la patología distrófica se evitó en gran medida y los autores no examinaron ninguna posible reversión del fenotipo de la enfermedad.
Por último, los autores compararon los resultados inmunológicos de la administración de un AAV-µDys con los del AAV-µUtro en el perro pointer alemán de pelo corto, que tiene un gen de la distrofina totalmente eliminado. Los perros GRMD tienen una pequeña cantidad de distrofina expresada como resultado de la lectura de la mutación y, por lo tanto, no son una buena prueba de inmunogenicidad. Observaron una fuerte respuesta inmunológica sistémica mediada por células a AAV-CMV-µDys pero no a AAV-CMV-µUtro. En ambos estudios se utilizó el promotor del CMV activo para impulsar la expresión de los transgenes. Será importante observar si la inmunogenicidad de la distrofina se ve cuando se impulsa desde los promotores específicos del músculo que se están probando actualmente en la clínica.
Ni la utrofina de longitud completa ni la µUtro pueden asociarse con la nNOS11, una proteína importante para regular el flujo sanguíneo a los músculos que se ejercitan, pero esta falta de asociación puede ser compatible con la mejora de la patología, ya que muchos individuos con BMD leve carecen de este dominio de localización de la nNOS6. Otras diferencias entre la distrofina y la utrofina pueden no ser importantes para el beneficio clínico. Además, la eficacia funcional de AAV-µUtro en el corazón no se probó en este estudio, pero el promotor del CMV sí permitió la expresión cardíaca. Se ha aprendido mucho de la labor de AAV-µDys, pero Song y sus colaboradores presentan una nueva y apasionante oportunidad de restablecer potencialmente la función normal en personas con DMD sin una posible respuesta inmunológica, lo que podría simplificar la repetición de la administración, si fuera necesario. Además, como la utrofina y la distrofina pueden estar localizadas conjuntamente en la membrana, los individuos con DMD también podrían beneficiarse. Aunque los actuales ensayos de terapia génica para la DMD están mostrando resultados alentadores, no se ha realizado una prueba rigurosa de la inmunidad a la distrofina, y la disponibilidad de vectores de µ-Utro proporciona un importante enfoque alternativo para tratar este devastador trastorno genético.
Fuente bibliográfica
Surrogate gene therapy for muscular dystrophy
Kay E. Davies & Jeffrey S. Chamberlain
MDUK Oxford Neuromuscular Centre, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK.
Nat Med 25, 1473–1474 (2019).