Intervención génica para la musculatura
El sistema de edición génica CRISP-Cas9 tiene enormes ventajas que pueden ser utilizadas para superar patologías de índole genético, como lo es la distrofia muscular. En estos casos, una mutación en una región codificante específica del gen de la proteína distrofina, impide tener una correcta función muscular. Recientes investigaciones realizadas en modelos animales para el estudio de esta patología, han otorgado valiosos resultados: se ha logrado escindir la secuencia mutada que genera la anomalía, mediante un sistema de entrega de genes necesarios para la edición molecular del gen alterado. Estos prometedores resultados fomentan la esperanza para que a futuro, estos enfoques puedan ser traducidos en terapias efectivas para combatir enfermedades tan invalidantes.
Distrofia muscular de Duchenne
El método de ingenería genética más revolucionario en estos días es el sistema CRISPR-Cas9, que recientemente se ha utilizado para tratar ratones con una frecuente patología: la distrofia muscular de Duchenne (DMD). Tres grupos de investigadores han descrito recientemente el uso del sistema de edición génica CRISPR-Cas9 para eliminar una mutación en el gen codificante para la proteína distrofina (DMD) y de esta forma modificar la expresión de la proteína.
Mediante el sistema CRISPR-Cas9, lograron cortar la parte mutada de la DMD en el modelo de roedores mdx (ocupado para estudiar la patología) posiilitando la síntesis de una forma proteica más corta en las fibras musculares, pero suficiente para la restauración parcial de la función muscular (fig. 1). Este tipo de enfoque podría eliminar mutaciones propias de la complicación en aproximadamente el 80% de los pacientes con DMD, aunque requeriría un mayor conocimiento sobre las secuencias específicas de cada mutación presente en cada afectado, y por ende, una estrategia personalizada para su eliminación.
Los autores entregaron todos los componentes necesarios del complejo CRISPR-Cas9 al músculo de los animales, utilizando vectores virales adeno asociados (AAV), un vehículo de suministro génico eficaz para el músculo de ratón, pero que tiene capacidad limitada para entregar material genético (es decir, sólo se puede administrar ADN de tamaño inferior a 4,7 kb). El gen que codifica la endonucleasa Cas9 también se dispuso en un vector de AAV. La función de esta enzima es degradar el ADN, para lo que adicionalmente, se dispuso de secuencias de ARN guías (gRNA) incorporados en un segundo vector viral. Los gRNAs guían a CRISPR-Cas9 hacia secuencias específicas del genoma. Una vez que los gRNAs han unido sus secuencias complementarias de ADN, la endonucleasa digiere los nucleótidos de interés.
Los vectores AAV fueron suministrados a animales mdx, organismos modelo para estudiar la DMD. Los roedores llevan una mutación sin sentido presente en el exón 23 de la secuencia codificante para la proteína distrofina. Tal mutación resulta en un término prematuro de la traducción del ARN mensajero (ARNm) de la DMD, impidiendo la síntesis proteica. Utilizando la estrategia de corrección génica, los gRNA guían a la enzima Cas9 para que digiera dos intrones que flanquean al exón 23 (fig. 1). Los extremos cortados se ensamblan por la acción del mecanismo de reparación celular, denominado unión de extremos no homólogos, lo que permite que el marco de lectura del ARNm de Dmd permanezca intacto.
La distrofina pudo ser sintetizada, sin los aminoácidos codificados por el exón extirpado. La eficacia inicial de esta edición génica se estimó en alrededor del 2%. Sin embargo, los niveles de distrofina aumentaron con el tiempo, tal vez debido a una continua corrección, ya que los constructos moleculares de AAV permanecen presentes en las fibras musculares durante muchos meses. Un grupo de investigadores (Science. 2016 Jan 22;351(6271):407-11) presentaron evidencias sobre cómo la edición génica también se lleva a cabo en células satélite, las células madre residentes del músculo. La reparación en este tipo celular podría ser un valioso resultado, dado que pueden continuar produciendo fibras con una función completa. Una estrategia similar, pero que utiliza vectores adenovirales o electroporación para introducir CRISPR-Cas9 y los gRNA (diseñados para digerir los exones 21 al 23 Dmd) a los músculos de ratones mdx también proporciona evidencia sobre mejoras de la función muscular. Sin embargo, estos métodos de entrega son más difíciles de traducir a la clínica.
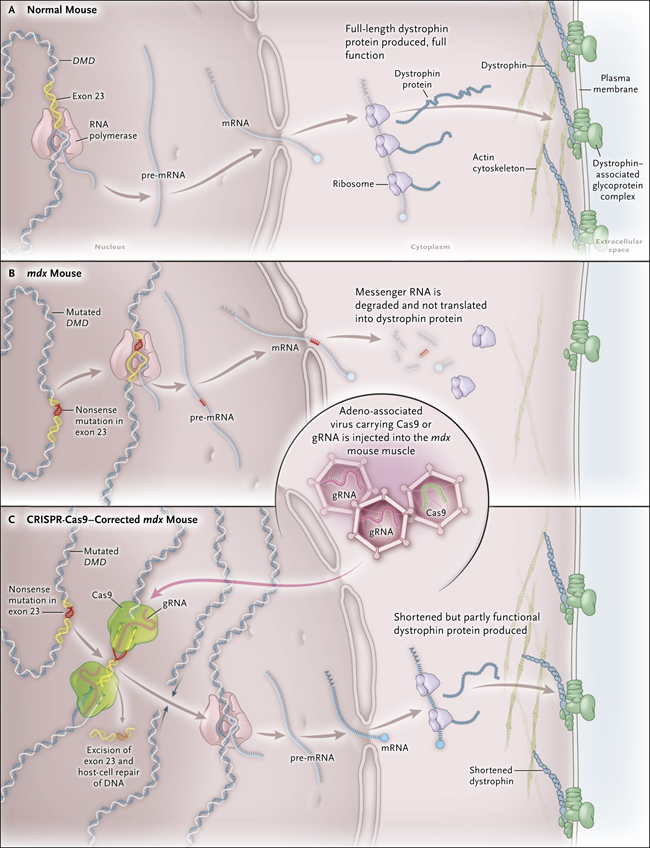
Figura 1. Estrategia de corrección mediante CRISPR-Cas9 en un modelo animal de distrofia muscular de Duchenne.
El gen que codifica la proteína distrofina se encuentra mutado en la distrofia muscular de Duchenne (DMD). La distrofina es una parte vital de complejos glicoproteicos que conectan el citoesqueleto de actina de una fibra muscular con la matriz extracelular para lograr la estabilización de la fibra muscular. Los ratones normales producen la proteína distrofina de longitud completa y sin alteraciones funcionales (panel A). En el ratón mdx, se produce un término anticipado en la síntesis de la proteína, debido a una mutación sin sentido en el exón 23, dando como resultado una nula producción de distrofina, y por ende ninguna funcionalidad (Panel B). Con el mecanismo de edición CRISPR-Cas9, dos ARN guía (gRNA) flanquean al exón 23 donde las endonucleasas Cas9 generan cortes en el ADN genómico. Los extremos cortados se vuelven a unir por enzimas de reparación celulares, proceso que elimina la secuencia alterada, permitiendo la generación de una distrofina corta pero que conserva parcialmente su función (panel C).
¿Cómo se compara el enfoque de CRISPR-Cas9 con otros métodos en desarrollo? Con la nueva tecnología, realizando un cambio en el genoma, se obtiene una solución permanente en las células musculares. Esto contrasta con el método de fármacos oligonucleotídicos que facilitan un "salto exónico" (exon-skipping), actualmente evaluados en ensayos clínicos. Estos oligonucleótidos fuerzan un splicing alternativo de tal manera que los exones que contienen la mutación son empalmados fuera del ARNm, dando como resultado la síntesis de una distrofina truncada.
Pero debido a que estas moléculas de omisión exónica no cambian la secuencia de DMD, tienen que ser readministrados frecuentemente. Otro enfoque involucra una terapia génica AAV con una versión "micro" truncada de DMD. La secuencia de ADN más corta se ajusta en un vector de AAV y tiene una eficacia considerable en ratones, aunque la síntesis de la proteína de longitud completa es más deseable. Esta estrategia no requiere la manipulación del genoma y además cada célula muscular que recibe el vector AAV - micro distrofina produce la proteína terapéutica. Por otra parte, la estrategia es de carácter general y no necesita ser personalizada.
Sin embargo, la expresión lograda a través del vector no es permanente, por lo que la síntesis de distrofina puede no ser tan duradera como la conseguida en células musculares corregidas mediante CRISPR. Los obstáculos para este nuevo enfoque, incluyen una eficacia inferior a la de otros métodos que se han propuesto. La expresión de Dmd es "rescatada" en sólo una fracción de las células tratadas. Aunque la precisión de CRISPR-Cas9 sigue mejorando, existe el riesgo de modificaciones fuera de objetivo en regiones del genoma. Por lo tanto, se requiere de trabajo adicional antes que CRISPR pueda ser considerado lo suficientemente seguro para su aplicación clínica. También existe el problema general de cómo CRISPR-Cas9 puede ser administrado eficientemente a la gran masa muscular implicada en la DMD.
Finalmente, el vector AAV funciona bien en ratones, pero la funcionalidad práctica para la entrega de cargas genéticas a grandes cantidades de tejido humano, aún se desconoce. Para lograr este objetivo, se necesitaría una gran cantidad de vectores y por lo tanto sería una fabricación difícil y costosa. Por otra parte, la respuesta inmunitaria contra la cápside de AAV, la proteína Cas9 y la misma distrofina, representa un potencial obstáculo. A pesar de esto, existen soluciones para estos problemas, así como enfoques alternativos que podrían evitarlos. La creciente aparición de novedosos métodos podrían conducir a remediar trastornos tan devastadores como la DMD y de todas formas representan un buen augurio para un exitoso final.
Fuente bibliográfica
The CRISPR Way to Think about Duchenne’s
Michele P. Calos, Ph.D.
Department of Genetics, Stanford University, Stanford, CA.
DOI: 10.1056/NEJMcibr1601383